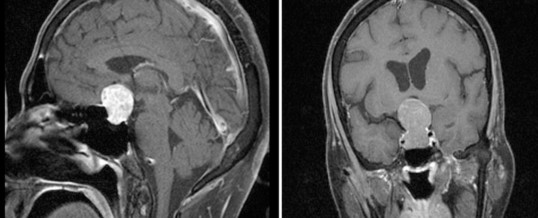
SINDROME DI NELSON
La sindrome di Nelson (Nelson’s syndrome) è una condizione che si sviluppa, non di rado, nei pazienti precedentemente affetti da malattia di Cushing sottoposti ad asportazione di entrambi i surreni (surrenectomia bilaterale).
Essa consiste nel rapido e progressivo sviluppo di una lesione espansiva a livello ipofisario che origina da cellule ipofisarie residuate dopo l’intervento di asportazione dell’adenoma ACTH-secernente che aveva determinato la malattia di Cushing.
I meccanismi fisiopatologici alla base della sindrome di Nelson non sono ancora perfettamente noti ed i fattori che portano al suo sviluppo sono ancora oggetto di dibattito. Tuttavia si ritiene probabile che la surrenectomia, correggendo l’ipercortisolismo, elimina il feedback negativo sull’ipotalamo, determinando un aumento della produzione ipotalamica di CRH. Di conseguenza i corticotropi ipofisari vengono iperstimolati con conseguente formazione di un voluminoso espanso ipofisario che caratterizza la sindrome di Nelson.
Il primo caso è stato descritto da Don Nelson nel 1958. Si trattava di una donna di 33 anni, sottoposta a surenectomia bilaterale per malattia di Cushing refrattaria alle terapie, che aveva sviluppato iperpigmentazione, difetti visivi, cefalea (prenota una visita neurologica).e presentava valori molto elevati di ACTH. Da allora i casi di sindrome di Nelson pubblicati sono decisamente aumentati. (Prenota una visita endocrinologica).
EPIDEMIOLOGIA
La malattia di Cushing è una patologia rara. Tuttavia, nei pazienti affetti da malattia di Cushing sottoposti a surrenectomia bilaterale, la sindrome di Nelson può svilupparsi non infrequentemente. Si ritiene che la sindrome di Nelson avvenga nel 8-43% degli adulti e nel 25-66% dei bambini.
Sebbene la sindrome di Nelson compaia mediamente 15 anni dopo la surrenectomia, sono decritti casi sviluppatisi anche dopo 24 anni e per questo motivo è necessario un monitoraggio del paziente anche a lungo termine.
La mortalità de sindrome di Nelson inizialmente riportata era discretamente elevata (12%). Oggi, tuttavia, si ritiene essa sia decisamente inferiore. (Prenota una visita endocrinologica).
DIAGNOSI
La diagnosi di sindrome di Nelson si basa su alcuni aspetti clinici, ormonali e radiologici.
Per ciò che riguarda l’aspetto clinico in passato, dato che la diagnosi veniva posta più tardivamente e l’adenoma ipofisario aveva più tempo per crescere di dimensioni, prevalevano alcuni sintomi o segni legati alla presenza di un’importante lesione espansiva nel cavo sellare quali: disturbi del campo visivo e paralisi dei nervi cranici). Oggi, invece, in virtù dell’esistenza di mezzi diagnostici più efficaci la diagnosi viene posta più precocemente. Pertanto i primi segni/sintomi che si riscontrano sono solitamente: cefalea, diabete insipido, panipopituitarismo, dolore testicolare, oligospermia; più raramente la sindrome di Nelson si associa a tumori paraovarici o paratesticolari come conseguenza dell’iperstimolazione degli adrenal rests nelle gonadi. Solo tardivamente è possibile riscontrare disturbi del campo visivo e paralisi dei nervi cranici.
Un segno caratteristico quasi costantemente presente è l’iperpigmentazione della cute, delle mucose, specialmente nelle superfici di contatto, di estensione e di flessione e sulle cicatrici). Tale fenomeno è conseguente all’iperproduzione di alfa-MSH, derivato della proopiomelanocortina (POMC), che stimola i melanociti a produrre un pigmento (melanina) che detemina l’inscurimento cutaneo.
Il sospetto clinico, deve essere confermato anche da parametri di laboratorio (incremento dei valori di ACTH) e dalla radiologia (lesione ipofisaria in aumento dimensionale). (Prenota una visita neurochirurgica).
Infatti l’ACTH tende ad aumentare dopo l’asportazione dei surreni, per cui andrebbe sempre dosato nell’immediato post-intervento di surrenectomia. Inoltre, è importante che il prelievo per il dosaggio dell’ACTH venga effettuato al mattino (h 8:00), almeno 20 ore dopo l’ultima assunzione della terapia sostitutiva corticosteroidea e prima dell’assunzione della terapia del mattino.
L’imaging ipofisario viene effettuato mediante la risonanza ipofisaria con mezzo di contrasto (gadolinio) che conferma la presenza di una lesione ipofisaria in progressivo aumento dimensionale. E poichè il 20% dei Nelson si sviluppa nel primo anno ed il 35% entro il secondo anno dalla surrenectomia i controlli andrebbero effettuati dopo 3 mesi dalla surrenectomia e successivamente ogni 6 mesi per i primi due anni. (Prenota una visita endocrinologica).
In conclusione per porre diagnosi di sindrome di Nelson si ritiene che in un paziente con pregressa malattia di Cushing, sottoposto a surrenectomia bilaterale debba esser presente almeno uno dei seguenti criteri:
– lesione ipofisaria in progressiva espansione (in confronto con la risonanza pre-intervento di surrenectomia);
– valori di ACTH > 500 ng/l in progressivo aumento in almeno 3 controlli successivi (per aumento si intende un incremendi di almeno il 30% rispetto al primo controllo post-intervento di surrenectomia).
{kind=link}
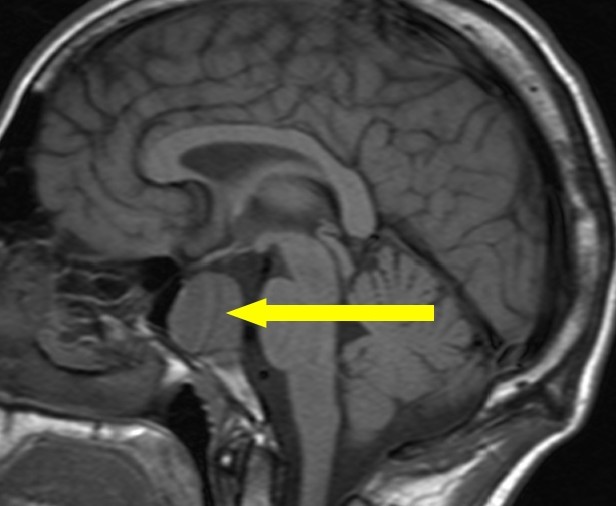{kind=link}
CAUSE
Il meccanismo fisiopatogenetico alla base della sindrome di Nelson non è ancora del tutto chiarito.
Tuttavia, è probabile che l’asportazione dei surreni, correggendo l’ipercortisolismo, elimini il feedback negativo sull’ipotalamo, determinando un aumento della produzione ipotalamica di CRH. Di conseguenza aumenterebbe la sintesi di proopiomelanocortina (POMC) e lo stimolo dei corticotropi ipofisari con conseguente crescita di un espanso ipofisario e sviluppo della sindrome di Nelson. (Prenota una visita endocrinologica).
FATTORI DI RISCHIO
Non è ancora del tutto chiaro quali siano tutti i fattori causali maggiormente responsabili dello sviluppo di una sindrome di Nelson.
Tuttavia, gli indici maggiormente predittivi di sviluppo di Nelson attualmente dimostrati sono i seguenti:
– la presenza di residuo tumorale all’imaging effettuato subito dopo l’intervento di asportazione dell’adenoma ipofisario;
– sottotipo aggressivo di corticotropinoma (definito in base alla velocità di crescita ed alla invasività della lesione);
– assenza di radioterapia ipofisaria neoadiuvante;
– rapido incremento dei valori di ACTH nel primo anno;
Non è escluso che altri fattori possano avere un ruolo nella genesi della sindrome di Nelson (i.e. durata della malattia di Cushing, età, presenza di residui surrenalici, entità dell’ipercortisolismo, insufficiente terapia steroidea sostitutiva, mancanza di soppressione del cortisolo al test di soppressione con alte dosi di desametasone), tuttavia le evidenze scientifiche in merito non sono ancora sufficienti. (Prenota una visita endocrinologica).
ANATOMIA PATOLOGICA
Le evidenze anatopatologiche hanno dimostrato che l’adenoma della malattia di Cushing e quello della sindrome di Nelson originano verosimilmente dallo stesso corticotropo.
E’ probabile, tuttavia, che alcune caratteristiche di maggior aggressività (ki-67, percentuale di mitosi, carcinoma corticotropo) dell’adenoma ACTH-secernente possano indicare o meno una maggior predisposizione allo sviluppo di Nelson.
TERAPIA
La terapia d’elezione è ovviamente la chirurgia. (Prenota una visita neurochirurgica). L’asportazione della massa ipofisaria può essere effettuata per via transnasosfenoidale o nei casi di Nelson con sviluppo sovrasellare può essere necessario l’approccio transcranico. La percentuale di successo varia dal 30 al 70%.
Nei casi residui di pazienti con sindrome di Nelson operati in cui invece si assite ad una progressione della malattia può essere utile la radioterapia neoadiuvante (tradizionale o con gamma-knife se la lesione è ben evidenzibile) o una terapia farmacologica.
Fra le terapie farmacologiche proposte suscita interesse un analogo della somatostatina di recente sviluppo (pasireotide o SOM230) che ha un’affinità per molti dei sottotipi recettoriali del recettore della somatostatina (SSTRs 1,2,3,5). Tuttavia per questa, come per le altre terapie farmacologiche proposte (PPAR-gamma, cabergolina, temozolomide, valproato di sodio) sono ancora necessari studi che ne confermino l’efficacia sugli umani affetti da Nelson.
Prenota una visita specialistica endocrinologica in merito a questo argomento.
Dott. Massimiliano Andrioli
Specialista in Endocrinologia e Malattie del Ricambio
Centro EndocrinologiaOggi, Roma
viale Somalia 33A, Roma
tel/fax 0686391386
cell 3337831426
Studio EndocrinologiaOggi, Lecce
via Ruffano 4, Casarano (Lecce)
tel/fax 0686391386
cell 3337831426
Bibliografia
1) TM Barber, E Adams, O Ansorge, JV Byrne , N Karavitaki, JAH Wass. Nelson’s Sindrome. European Journal of Endocrinology (2010) 163 495–507
2) Nelson DH, Meakin JW, Dealy JB, Matson DD, Emerson K & Thorn GW. ACTH-producing tumor of the pituitary gland. New England Journal of Medicine 1958 259 161–164.
3) Nelson DH, Meakin JW & Thorn GW. ACTH-producing pituitary tumors following adrenalectomy for Cushing’s syndrome. Annals of Internal Medicine 1960 52 560–569. 3 Nagesser SK, van Seters AP, Kievit J, Hermans J, Krans HM & van de Velde CJ. Long-term results of total adrenalectomy for Cushing’s disease. World Journal of Surgery 2000 24 108–113.
Share
NOV
2011