CARCINOMA IPOFISARIO
Il carcinoma ipofisiario è un tumore che insorge dalle cellule della regione anteriore dell’ipofisi (adenoipofisi) e che, per definizione, si associa a metastasi cerebrospinali o sistemiche.
Epidemiologia
Si tratta di una neoplasia molto rara che colpisce gli adulti, senza predilezione di sesso. In più del 75% le neoplasie sono ormono-secernenti. In ordine decrescente di frequenza gli ormoni prodotti sono: PRL (prolattinomi), ACTH (malattia di Cushing), GH (acromegalia) e TSH (TSHomi).
Eziologia
L’eziologia del tumore è sconosciuta e non sono note specifiche alterazioni geniche predisponesti allo sviluppo di carcinoma ipofisario. Non è chiaro, inoltre, se il carcinoma insorga ex novo da un’ipofisi normale, se è conseguenza di una trasformazione maligna di un preesistente adenoma ipofisario benigno o se si possano verificare entrambe le condizioni.
Clinica
La presentazione clinica del carcinoma dell’ipofisi è simile a quella dell’adenoma ipofisario che attualmente è stato rinominato PitNET. Pertanto si può associare ad un quadro clinico variabile in base all’ormone prodotto (iperprolattinemia, malattia di Cushing, acromegalia, ipertiroidismo). Più raramente la neoplasia è silente in quanto non produce ormoni (adenoma non secernente) e di conseguenza non si associa a sindromi cliniche (prenota una valutazione neurochirurgica).
Talvolta può accadere che un tumore diagnosticato come benigno dall’istologia possa manifestare metastasi dopo alcuni anni, in media 5 dopo nei prolattinomi e 10 anni dopo nei tumori che producono ACTH (malattia di Cushing).
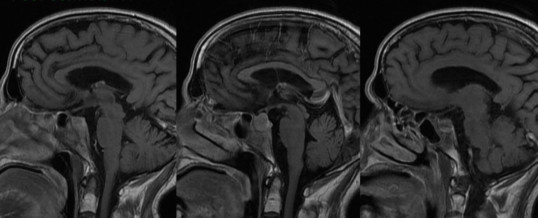
Il carcinoma ipofisario insorge solitamente nella sella turcica, le cellule neoplastiche proliferano velocemente ed assumono un carattere “infiltrativo”, espandendosi nelle strutture circostanti (osso e cervello). Le metastasi si localizzano nello spazio cranio-spinale o a distanza (fegato, polmoni, ossa e linfonodi).
Istologia
Macroscopicamente un carcinoma ipofisario è del tutto simile all’adenoma (figura A), ma può presentare piccoli noduli sulla parete ossea, sul tessuto subaracnoideo, sui nervi cranici o sulle meningi. Talvolta, già al momento della diagnosi possono esser presenti metastasi a distanza.
Dal punto di vista istologico, il reperto è sovrapponibile a quello dell’adenoma ipofisario (figura B). Si osserva una proliferazione di cellule polimorfe (diverse l’una dall’altra), con mitosi e necrosi (segno di maggiore aggressività) e possibile invasione delle strutture vicini (osso, cervello, meningi). Nei casi sospetti in cui manca l’evidenza clinica di metastasi, può esser utile valutare l’indice di proliferazione, che corrisponde alla percentuale delle cellule che si stanno moltiplicando in una neoplasia. Se l’indice di proliferazione è inferiore al 3%, la neoplasia è sicuramente benigna (adenoma). Se invece l’indice è superiore al 3% è più probabilmente che si tratti di un carcinoma ipofisario. L’indice di proliferazione è calcolato con un anticorpo che colora le cellule che stanno proliferando, chiamato Mib-1 o Ki 67 (indici di proliferazione) (figura C). Nel caso di ipersecrezione ormonale è possibile evidenziare anche la presenza di questi ormoni nelle cellule tumorali con anticorpi specifici che si legano a cellule specifiche (figura D).
Terapia
Per gli adenomi ipofisari rapidamente invasivi ed i carcinomi ipofisari resistenti alle terapie mediche i trattamenti chirurgici, radioterapici ed i comuni chemioterapici sono poco efficaci (prenota una valutazione neurochirurgica).
Si ritiene che l’agente alchilante Temozolomide possa essere un’opzione efficace in questi casi. La temozolomide è un agente citotossico alchilante, la cui citotossicità è dovuta principalmente a metilazione del DNA nelle posizioni N7 e O6 della guanina, con conseguente inibizione della replicazione del DNA. Questa metilazione determina rotture del DNA morte cellulare per apoptosi. Il danno viene generalmente corretto dall’enzima riparatore del DNA Metil-Guanil-Metil-Transferasi (MGMT). Le cellule tumorali che hanno bassi livelli di MGMT sono più sensibili alla citotossicità di temozolomide. Ne consegue che la metilazione del promotore del gene dell’MGMT si associa ad una migliore risposta terapeutica al farmaco. Tuttavia, sono possibili anche altri meccanismi d’azione alternativi.
La temozolomide si somministra per via orale ed è stata recentemente autorizzata nel trattamento degli adenomi aggressivi o carcinomi ipofisari. Il dosaggio previsto è di 200 mg/m2 per 5 giorni ogni 28 giorni. La durata del trattamento è in genere di 6 mesi.
Prenota una visita specialistica endocrinologica in merito a questo argomento.
Dott.ssa Paola Parente
e
Dott. Massimiliano Andrioli
Specialista in Endocrinologia e Malattie del Ricambio
Centro EndocrinologiaOggi, Roma
viale Somalia 33A, Roma
tel/fax 0686391386
cell 3337831426
Studio EndocrinologiaOggi, Lecce
via Ruffano 4, Casarano (Lecce)
tel/fax 0686391386
cell 3337831426
Share
GIU
2013