TSH-OMA (ADENOMA TSH SECERNENTE)
Il TSH-oma è un adenoma ipofisario secernente TSH derivante dalle cellule tireotrope dell’adenoipofisi.
Epidemiologia e Classificazione
Gli adenomi ipofisari possono essere classificati in base alle dimensioni, all’estensione, all’infiltrazione delle strutture adiacenti e alla capacità di sintetizzare e secernere ormoni. Attualmente sono stati rinominati PitNet.
Per quanto riguarda le dimensioni, gli adenomi con diametro <1 cm vengono definiti microadenomi, mentre quelli con diametro maggiore sono definiti macroadenomi.
In base alla capacità secretoria delle cellule adenomatose, gli adenomi vengono distinti in:
- Adenomi non secernenti o NFPA: rappresentano il 25% circa degli adenomi ipofisari;
- Adenomi secernenti.
Questi ultimi vengono , a loro volta, classificati in base all’ormone prodotto in:
- Secernenti prolattina o Prolattinomi: 40-50%
– Secernenti GH o GH-omi: 20-25%
– Secernenti ACTH o ACTH-omi: 8-10%
– Secernenti FSH/LH o Gn-omi: <1%
– Secernenti TSH: <1%
Gli adenomi ipofisari secernenti TSH o TSH-omi rappresentano una patologia rara con una prevalenza di circa 1 caso per milione di persone con rapporto M:F=1:1,5. Nell’ultima decade il numero di TSH-omi segnalati è triplicato, principalmente grazie all’introduzione della misurazione del TSH ultrasensibile. Sono stati riportati casi in soggetti di età compresa tra gli 8 e gli 84 anni, con un picco di insorgenza nella 5-6a decade di vita. Si presentano prevalentemente in forma sporadica ma sono stati descritti casi familiari di TSH-oma nell’ambito della sindrome neoplastica endocrina multipla di tipo 1 (MEN 1) e nell’adenoma ipofisario isolato familiare (FIPA) con mutazione AIP. Fino al 1996 più dell’80% dei casi segnalati era rappresentato da lesioni con diametro >1 cm, ma l’utilizzo di nuove metodiche di laboratorio ha contribuito, favorendo una diagnosi precoce, al progressivo aumento della prevalenza dei microadenomi. Fino a pochi decenni fa, quindi, la maggior parte degli adenomi secernenti TSH era rappresentata da macroadenomi invasivi con estensione extrastrellare. (Prenota una visita neurochirurgica).
Tali lesioni possono secernere tireotropina in forma isolata oppure in associazione ad altre tropine ipofisarie o alle subunità α degli ormoni glicoproteici:
– Secrezione isolata di TSH (70%)
– Secrezione mista TSH/GH (18%)
– Secrezione mista TSH/PRL (10%)
– Secrezione mista TSH/gonadotropine (<2%)
In letteratura sono stati descritti pochissimi casi di trasformazione carcinomatosa. Livelli molto elevati di subunità α libera associati ad una diminuzione spontanea e marcata dei livelli sierici di TSH potrebbero indicare che il tumore sta diventando meno differenziato e correlare con la sua invasività e tendenza a metastatizzare.
Clinica
Il quadro clinico relativo alla presenza di un adenoma ipofisario dipende dalle dimensioni e dai rapporti che lo stesso sviluppa con le strutture adiacenti e, nel caso degli adenomi secernenti, dalla sindrome ipersecretoria associata.
I segni e sintomi legati all’effetto massa dell’adenoma sono indipendenti dalla capacità secretoria e sono causati dalla compressione ed infiltrazione delle strutture contigue esercitata dalle cellule adenomatose.
La tendenza all’invasività e la frequente estensione sopra-sellare riscontrata nei TSH-omi aumentano la probabilità che la sintomatologia clinica legata all’effetto massa sia manifesta. Possono essere presenti alterazioni del campo visivo (quadrantanopsia superiore/emianopsia bitemporale/scotoma centrale monolaterale) (prenota un campo visivo) per compressione del chiasma ed eventuale successivo coinvolgimento del nervo ottico, oftalmoplegia o diplopia per compressione dei nervi cranici a livello del seno cavernoso, idrocefalo per compressione del terzo ventricolo e occlusione dei forami di Monro e rinoliquorrea per erosione del pavimento sellare con infiltrazione nel seno sfenoidale (prenota una visita oculistica). La cefalea frontale, continua e resistente agli analgesici, causata dalla distensione del diaframma sellare, è un sintomo presente nel 20-25% dei casi, e può essere associata, più raramente, ad altri sintomi e segni di ipertensione endocranica come il vomito e l’edema della papilla per effetto della massa espansiva intracranica (prenota un visita neurologica). L’effetto compressivo delle cellule adenomatose sulle cellule adeno-ipofisarie circostanti può determinare alterazioni secretorie a carico di queste ultime che possono condurre ad un quadro di ipopituitarismo, presente in circa ¼ dei pazienti. La diversa sensibilità delle cellule dell’adeno-ipofisi all’effetto compressivo stabilisce l’ordine progressivo secondo il quale si manifesta il deficit secretorio: vengono colpite prima le cellule secernenti GH, poi quelle secernenti gonadotropine, successivamente quelle TSH-secernenti ed infine quelle corticotrope. A causa della compressione del peduncolo ipotalamo-ipofisario con perdita dell’inibizione dopamino-mediata sulla secrezione di prolattina, può essere riscontrata pseudoiperprolattinemia. La compressione del peduncolo può causare, inoltre, diabete insipido per interruzione del flusso assonico che trasporta gli ormoni neuro-ipofisari. Sono stati descritti rari casi di esoftalmo unilaterale dovuto ad invasione orbitale da parte dell’adenoma ipofisario, da porre in diagnosi differenziale con l’esoftalmo tipico dell’orbitopatia a genesi autoimmune (prenota un visita oculistica).
A questi segni e sintomi, vanno aggiunti quelli legati alla secrezione inappropriata di TSH. L’espressione ridotta o assente di recettori per gli ormoni tiroidei sulle cellule adenomatose TSH-secernenti rende la secrezione di TSH totalmente o parzialmente svincolata dal feedback inibitorio esercitato dagli ormoni tiroidei stessi sulla sintesi e secrezione della tireotropina. Pertanto, Il TSH può esercitare in modo abnorme la sua azione stimolatoria sul trofismo e sulla funzione dei tireociti, portando allo sviluppo del gozzo, sempre presente anche nella sua variante multinodulare (72%), e di sintomi e segni legati all’ipertiroidismo, in genere più sfumati di quanto farebbero presupporre gli elevati valori ormonali. Possono essere presenti tachicardia, tremori, intolleranza al caldo ed astenia, alvo frequente ed irritabilità.
In letteratura, inoltre, diversi pazienti non trattati sono stati descritti come clinicamente eutiroidei: la secrezione di molecole di TSH con scarsa attività biologica potrebbe rappresentare la peculiarità di questi TSH-omi “silenti”.
In caso di secrezione mista, alla sintomatologia legata all’ipersecrezione degli ormoni tiroidei, si associa quella legata all’ipersecrezione delle altre tropine ipofisarie.
Diagnosi
In caso di segni e sintomi caratteristici di ipertiroidismo, è necessario valutare la funzionalità tiroidea dosando TSH, FT3, FT4, che risulteranno tipicamente elevati permettendo l’esclusione di tutte le forme di ipertirodismo primario.
Tuttavia, circa il 30% dei pazienti con TSH-oma e tiroide intatta mostra livelli di TSH entro il range di normalità: la secrezione di molecole di TSH con aumentata attività biologica potrebbe spiegare tale evidenza. Le molecole di TSH secrete dai tumori ipofisari sono eterogenee e possono presentare attività biologiche e immunologiche normali, ridotte o aumentate, probabilmente in seguito alla modificazione dei processi di glicosilazione secondaria alle alterazioni dell’elaborazione post-traduzionale dell’ormone all’interno della cellula tumorale.
L’ecografia tiroidea evidenzierà la presenza di gozzo semplice o più frequentemente multinodulare (prenota un’ecografia tiroidea).
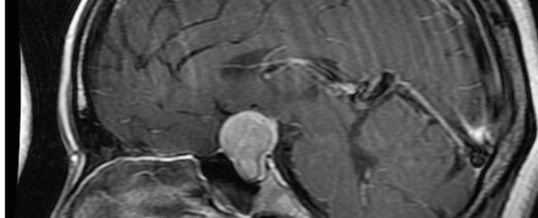
Nel sospetto clinico di adenoma ipofisario, lo studio morfologico della regione sellare mediante RMN con e senza mezzo di contrasto permetterà di individuare la lesione qualora presente, valutarne le dimensioni e i rapporti con le strutture circostanti. La scintigrafia ipofisaria con octreotide radiomarcato (octreoscan) ha dimostrato di localizzare con successo i TSH-omi che esprimono i recettori della somatostatina; tuttavia, la specificità dell’octreoscan è bassa. In presenza di adenoma ipofisario, è necessario valutare la funzione ipotalamo-ipofisaria dosando le restanti tropine ipofisarie (PRL, GH, ACTH, LH e FSH). I pazienti con macroadenoma devono essere sottoposto a videat neuro-oftalmologico per la valutazione dell’acuità visiva e per l’esecuzione della campimetria (prenota campimetria) e dell’esame del fundus oculi (prenota fondo oculare).
Tenendo sempre presente la possibilità di imbattersi in un incidentaloma ipofisario, è necessario considerare che l’altra principale causa di inappropriata sintesi di TSH è la resistenza agli ormoni tiroidei RTHβ, che nella sua forma selettiva ipofisaria si manifesta con sintomi e segni tipici di ipertiroidismo, rendendo necessaria, nei casi dubbi (adenoma ipofisario di dimensioni estremamente ridotte o alterazioni confondenti come la empty sella), una diagnosi differenziale tra la sindrome da resistenza ipofisaria e l’adenoma ipofisario TSH secernente mediante i seguenti approfondimenti diagnostici:
- Test di soppressione con T3, in cui, in caso di TSH-oma, non si verifica soppressione del TSH;
- Test di stimolo con TRH, che non mostra aumento del TSH nei pazienti affetti da TSH-oma;
- Dosaggio della subunità α del recettore degli ormoni tiroidei, che risulta aumentata in caso di TSH-oma, con un rapporto subunità α/TSH >5,7
Inoltre, la valutazione dei marcatori metabolici dell’azione degli ormoni tiroidei può essere utile nella diagnosi differenziale, dimostrando valori aumentati degli stessi in caso di TSH-oma.
Inoltre, nei casi problematici di ipertiroidismo di origine centrale, è stata proposta da alcuni autori la somministrazione di analoghi della somatostatina a lunga durata d’azione per almeno 2 mesi per effettuare una diagnosi differenziale: solo i pazienti con TSH-oma risultano responsivi a tale trattamento mostrando una rapida riduzione dei valori di TSH.
Terapia e Controllo
L’approccio terapeutico è correlato alla localizzazione e all’estensione dell’adenoma e alla sindrome ipersecretoria eventualmente presente.
La terapia è basata sull’associazione tra chirurgia, radioterapia e terapia farmacologica.
La chirurgia rappresenta l’opzione terapeutica di prima scelta con l’obiettivo di rimuovere il tessuto neoplastico e ripristinare la normale funzione pituitaria e tiroidea; sono possibili due approcci chirurgici in base al volume tumorale e al suo grado di estensione: transfenoidale e transcranico subfrontale.
Tuttavia, una rimozione radicale degli adenomi ipofisari secernenti TSH può risultare particolarmente difficoltosa a causa della marcata fibrosi che li caratterizza e dell’invasione locale che può coinvolgere le strutture vascolari e nervose circostanti.
Prima di affrontare l’intervento chirurgico (prenota valutazione neurochirurgica) è necessario ripristinare l’eutiroidismo mediante l’utilizzo di analoghi della somatostatina (OCTREOTIDE 100 mcg 3 volte/die sc – OCTREOTIDE LAR 10-30 mg ogni 4 settimane – LANREOTIDE) che sono risultati efficaci nel ridurre la secrezione del TSH in più del 90% dei pazienti con conseguente normalizzazione degli ormoni tiroidei circolanti. Possono essere utilizzati in alternativa farmaci antitiroidei (METIMAZOLO 20-30 mg die – PROPILTIOURACILE 200-300 mg/die, per os) in associazione con beta-bloccanti (PROPANOLOLO 80–120 mg/die, per os).
E’ importante tenere presente che il ripristimo dell’eutiroidismo pre-operatorio elimina l’azione inibitoria esercitata dagli elevati livelli di ormoni tiroidei sulle cellule tireotrope normali; questa riattivazione può rendere difficoltosa la valutazione della funzionalità tiroidea nei primi giorni post-intervento, poiché, anche in caso di rimozione completa, il TSH potrebbe non risultare indosabile. Infatti, livelli di TSH non rilevabili una settimana dopo l’intervento possono indicare una rimozione completa solo a condizione che i trattamenti farmacologici vengano interrotti prima dell’intervento chirurgico (prenota una visita neurochirurgica).
Dopo l’intervento il TSH può permanere basso per settimane o mesi; il conseguente ipotiroidismo centrale va trattato con levotiroxina a dosaggio sostitutivo controllando periodicamente la funzionalità tiroidea.
La valutazione della funzionalità ipofisaria residua va effettuata nell’immediato post-intervento, con particolare attenzione alla valutazione dell’asse ipotalamo-ipofisi-surrene. Se l’ipopituitarismo è causato dalla compressione del peduncolo ipofisario e/o dei vasi portali, è possibile che si verifichi un recupero della funzione secretoria ipofisaria: la possibilità che possa verificarsi tale recupero può essere sondata mediante l’esecuzione pre-operatoria dei test di stimolo della funzione ipofisaria che mostrano una risposta normale se non vi è stata distruzione delle cellule adeno-ipofisarie.
Nei casi in cui la l’ablazione chirurgica risulti incompleta o qualora il paziente rifiuti l’intervento, la radioterapia ipofisaria e/o il trattamento medico con analoghi della somatostatina sono due valide alternative.
La radioterapia è indicata :
- Nelle forme secernenti con persistenza di ipersecrezione ormonale dopo intervento chirurgico e/o terapia farmacologica
- Nei residui di adenoma invasivo non trattabili chirurgicamente
- Dopo asportazione di recidiva dell’adenoma
Se l’adenoma è localizzato ad una distanza >3 mm dal chiasma o dai nervi ottici e presenta un diametro ≤25 mm, è preferibile optare per l’approccio radiochirurgico stereotassico gamma knife che consente di indirizzare in una sola seduta un’elevata dose di radiazioni ad una zona bersaglio; la dose raccomandata è di 10-25 Gy in dose singola e i risultati terapeutici sono apprezzabili entro tempi più brevi con una minore incidenza di effetti collaterali. Se il tumore è localizzato in prossimità del chiasma o dei nervi ottici, invece, è preferibile affidarsi alla radioterapia esterna convenzionale con somministrazione di una dose non inferiore a 45 Gy frazionati in 2 Gy al giorno, per 5 giorni a settimana per 5 settimane. Questo approccio garantisce un basso rischio di complicanze tardive alle strutture nervose, in particolare alle vie ottiche, ma necessita di tempi più prolungati per l’ottenimento dei risultati terapeutici ed è gravato da una maggiore incidenza di effetti collaterali. In più del 70% dei pazienti sottoposti a radioterapia si verifica guarigione completa. Tra le complicanze più frequenti del trattamento radioterapico, soprattutto con approccio esterno convenzionale, vi è la comparsa di ipopituitarismo, la cui prevalenza aumenta con la durata del periodo di controllo; per tal motivo, nei pazienti sottoposti a radioterapia, le tropine ipofisarie devono essere monitorate costantemente al fine di individuare con tempestività eventuali deficit.
Dopo l’irradiazione o in alternativa ad essa, il pz dovrà proseguire la terapia medica in attesa della normalizzazione dei valori ormonali.
I dopamino-agonisti, e in particolare la CABERGOLINA, sono stati impiegati in alcuni TSH-omi con risultati variabili: gli effetti positivi sono stati osservati principalmente in alcuni pazienti con adenoma a secrezione mista PRL/TSH.
Il trattamento medico degli adenomi TSH-secernenti si basa sull’utilizzo di analoghi della somatostatina a lunga durata d’azione (OCTREOTIDE LAR 10-30 mg ogni 4 settimane – LANREOTIDE) che porta ad una riduzione del TSH nella quasi totalità dei casi, con il ripristino dello stato eutiroideo e una riduzione volumetrica del gozzo nel 30% dei pazienti, e alla riduzione volumetrica dell’adenoma ipofisario in circa il 40% dei pazienti con miglioramento dei sintomi visivi nel 70% dei casi. La dose somministrata deve essere individualizzata in base alla risposta terapeutica.
In caso di fallimento dei suddetti approcci terapeutici ed in presenza di ipertiroidismo potenzialmente letale, si può ricorrere alla tiroidectomia chirurgica (prenota una visita chirurgica) e all’ablazione tiroidea mediante terapia radiometabolica, seguita dalla terapia con levotiroxina a dosaggio sostitutivo.
L’ablazione tiroidea è stata a lungo considerata con cautela poiché si sospettava potesse aumentare il rischio di trasformazione aggressiva dell’adenoma, come osservato nella sindrome di Nelson dopo surrenectomia per la malattia di Cushing. Questa argomentazione è stata supportata principalmente da due osservazioni: in primo luogo, i livelli di TSH in pazienti precedentemente sottoposti ad ablazione tiroidea sono risultati fino a sei volte superiori rispetto ai pazienti non trattati; in secondo luogo, nei pazienti trattati, è stata osservata una frequenza maggiore di macroadenomi invasivi. È stato, quindi, ipotizzato che le cellule adenomatose potessero mantenere almeno parzialmente la loro reattività ai meccanismi di feedback incrementando la secrezione di TSH e proliferando più attivamente in risposta alle riduzioni anche minime dei livelli circolanti dell’ormone tiroideo che possono verificarsi in caso di trattamenti ablativi coinvolgenti la tiroide. Contro questa ipotesi ci sono i risultati ben documentati dei test di soppressione con T3 a cui sono stati sottoposti i pazienti affetti da TSH-omi che dimostrano l’autonomia funzionale delle cellule adenomatose rispetto al feedback esercitato dell’ormone tiroideo.
In generale, il paziente deve essere valutato clinicamente e biochimicamente ogni 4-6 mesi per il primo anno dopo l’intervento, e poi ogni anno. L’imaging pituitario dovrebbe essere eseguito ogni 6 mesi nel primo anno, e successivamente dopo 2 anni in assenza di segni e sintomi di compressione locale. In caso di macroadenoma persistente, è opportuno eseguire un videat oculistico (prenota visita oculistica) con studio campimetrico (prenota campo visivo computerizzato) ogni anno o in seguito alla comparsa di alterazioni dell’acuità e del campo visivo.
È, infatti, importante sottolineare che la decompressione chirurgica può migliorare o normalizzare i deficit campimetrici solo nei casi in cui la compromissione del chiasma e/o del nervo ottico sia di recente insorgenza e non sia presente atrofia papillare (prenota visita oculistica).
Prenota una visita endocrinologica in merito a questo argomento.
Dott.ssa Silvia Carocci
e
Dott. Massimiliano Andrioli
Specialista in Endocrinologia e Malattie del Ricambio
Centro EndocrinologiaOggi, Roma
viale Somalia 33A, Roma
tel/fax 0686391386
cell 3337831426
Studio EndocrinologiaOggi, Lecce
via Ruffano 4, Casarano (Lecce)
tel/fax 0686391386
cell 3337831426
Bibliografia
- P. Beck-Peccoz, A. Lania, A. Beckers, K. Chatterjee, and J.-L. Wemeaue. 2013 European Thyroid Association Guidelines for the Diagnosis and Treatment of Thyrotropin-Secreting Pituitary Tumors. Eur Thyroid J. 2013 Jun; 2(2): 76–82.
- Lee W, Cheung AS, Freilich R. TSH-secreting pituitary carcinoma with intrathecal drop metastases. Clin Endocrinol (Oxf). 2012 Apr;76(4):604-6
- Daly AF, Tichomirowa MA, Petrossians P, et al. Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: an international collaborative study. J Clin Endocrinol Metab. 2010 Nov;95(11):E373-83.
- Beck-Peccoz P1, Persani L, Mannavola D, Campi I. Pituitary tumours: TSH-secreting adenomas. Best Pract Res Clin Endocrinol Metab. 2009 Oct;23(5):597-606.
- Losa M1, Fortunato M, Molteni L, Peretti E, Mortini P. Thyrotropin-secreting pituitary adenomas: biological and molecular features, diagnosis and therapy. Minerva Endocrinol. 2008 Dec;33(4):329-40.
- Daousi C, Foy PM, MacFarlane IA. Ablative thyroid treatment for thyrotoxicosis due to thyrotropin-producing pituitary tumours. J Neurol Neurosurg Psychiatry. 2007 Jan;78(1):93-5.
- Foppiani L, Del Monte P, Ruelle A, Bandelloni R, Quilici P, Bernasconi D. TSH-secreting adenomas: rare pituitary tumors with multifaceted clinical and biological features. J Endocrinol Invest. 2007 Jul- Aug;30(7):603-9.
- Brown RL, Muzzafar T, Wollman R, Weiss RE. A pituitary carcinoma secreting TSH and prolactin: a non-secreting adenoma gone awry. Eur J Endocrinol. 2006 May;154(5):639-43.
- Mannavola D1, Persani L, Vannucchi G, Zanardelli M, Fugazzola L, Verga U, Facchetti M, Beck-Peccoz P. Different responses to chronic somatostatin analogues in patients with central hyperthyroidism. Clin Endocrinol (Oxf). 2005 Feb;62(2):176-81.
- Caron P1, Arlot S, Bauters C, Chanson P, Kuhn JM, Pugeat M, Marechaud R, Teutsch C, Vidal E, Sassano P. Efficacy of the long-acting octreotide formulation (octreotide-LAR) in patients with thyrotropin-secreting pituitary adenomas. J Clin Endocrinol Metab. 2001 Jun;86(6):2849-53.
- Beck-Peccoz P1, Persani L. Medical management of thyrotropin-secreting pituitary adenomas. Pituitary. 2002;5(2):83-8.
- Caron P, Arlot S, Bauters C, Chanson P, Kuhn JM, Pugeat M, Marechaud R, Teutsch C, Vidal E, Sassano P. Efficacy of the long-acting octreotide formulation (octreotide-LAR) in patients with thyrotropin-secreting pituitary adenomas. J Clin Endocrinol Metab 2001
- Taylor TJ, Donlon SS, Bale AE, Smallridge RC, Francis TB, Christensen RS, Burma KD. Treatment of a thyrotropinoma with octreotide-LAR in a patient with multiple endocrine neoplasia-1. Thyroid 2000
- Beck-Peccoz P, Brucker-Davis F, Persani L, Smallridge RC, Weintraub BD. Thyrotropin-secreting pituitary tumors. Endocr Rev 1996; 17:610-638
- Gancel A, Vuillermet P, Legrand A, Catus F, Thomas F, Kuhn J M. Effets of a slow-release formulation of the new somatostatin analogue lanreotide in TSH-secreting pituitary adenomas. Clin Endocrinol (Oxf) 1994; 40:421-428
- “Endocrinologia Clinica” – Fabrizio Monaco (IV edizione)
GEN
2018